Twilight – that time between the clarity of day and the murky darkness of night.
Ehlers-Danlos Syndrome
When I got diagnosed with Ehlers-Danlos Syndrome (EDS) at 55, I was told by the doc that it was untreatable and uncureable and to just go home and forget about it. Once so much came online about it, I was able to get further but except for odd surgeries, cysts and hernias, it all seem to just go along without massive problems.
Just before and after a series of surgeries and my final colon removal, symptoms took a step up. At the time I just thought it was EDS getting worse – being activated by surgeries, infections, and/or antibiotics. I started bracing as my joints started hurting. I thought maybe it was mild subluxations even though I had never had a dislocation or many problems with joints.
Then we moved from Virginia to Maine – requiring several long drives, packing, unpacking etc. New symptoms appeared – cramping feet, toes, numbness/tingling like odd body parts were going to “sleep”. My extremely mild asthma got worse but normal for Maine since as the tailpipe of the nation, the state has higher rates of asthma. My GI distress continued to rise leading me to wonder if my gastroparesis was worse, did I have short colon syndrome though I had only lost 6 inches of the small intestines? I had to quit eating a number of things.
Mast Cell Activation Syndrome
Then came deep digs into Facebook groups for EDS. I started seeing more about Mast Cell Activation Syndrome (MCAS) and I decided to learn about it, joining other FB groups for the condition. I started actively researching across medical sites. Once the asthma worsened, I suggested to my doctor to try singular, a med I had been on in the past. Lo and behold my facial flushing diminished – a good indicator that I might have MCAS. Later that year I was able to find a doc (telemedicine in Colorado) to diagnose me with MCAS. That got my PCP moving and I subsequently got a referral to a prominent Boston MCAS doctor. Oh my, things improved! Some clarity, no longer twilight, I thought!
Well, the GI issues did improve. My pain level also improved: the joint pain was sign of MCAS reactions. The other symptoms, however, continued.
Next up: Myasthenia Gravis
Then I had an incident while driving, my eyes closed and my vision issues continued to worsen. I was able to get to the eye doc who prescribed drops for the dryness, and treatment for the blepharitis. The drops were painful and did not improve anything.
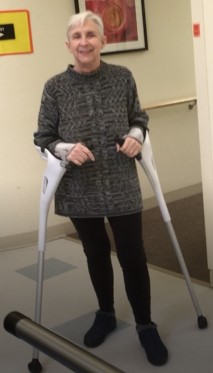
I started using forearm crutches to help keep me straighter and improve my dystonia which could make me look drunk. I was prescribed a back brace; I was able to stand straighter and it did help build my core muscles, destroyed after multiple abdominal surgeries. I have a genetic tendency towards kyphosis, hunchback, so I just thought I was hitting the same stage as my grandmother at the same age.
I also had two bouts of leg weakness – once after restarting cymbalta and once out of the blue. Since dysautonomia (mostly POTS) is prevalent among EDS patients, my PCP agreed to send me to a neurologist for autonomic testing. I knew I didn’t have POTS but also knew my autonomic system was definitely abnormal. I didn’t actually think the testing would highlight anything I didn’t know. I mean really folks, I stopped sweating entirely decades ago! The testing proved what I already knew but giving me a nebulous diagnosis of autonomic neuropathy. Not really very helpful.
That medical appointment introduced me to entirely new territory. Weeks later I lost the ability to open my eyes for several days. The neurologist had mentioned myasthenia gravis after he spotted my drooping eyes. Blood testing immediately showed none of the antibodies and then shortly afterwards my descent into darkness, literally and figuratively began.
It’s the pandemic. It’s the short staffing. It’s normal medical inability to reach a rare conclusion. It’s a neurologist who may not actually be that well-versed in this rare disease, myasthenia gravis.
That was October. Nearly February now and I have finally had my photosensitivity retreat back to my old normal. I still can’t read as along as I want and sometimes my eyes just won’t focus at all. I trialed a med which activated my MCAS and gave me horrible stomach cramping. My doc did not cooperate. My next appointment is mid-March.
The worse part of MG is the possibility of a crisis – when your chest stops working and you can’t breathe. This requires a hospital and sometimes even a ventilator. It can come on suddenly and too many medical personnel do not know about the condition. One’s blood oxygen level can still be 100% and you cannot breathe.
So I wait. I do not have the MG diagnosis in my records. My PCP is very conservative, very hesitant and wants directions from a MG specialist. Ha. And I wait. I document symptoms. I research. I become more convinced I have MG.
It is twilight. Heading towards darkness.
But I’ve been here before. EDS is a rare genetic condition. I actually am diagnosed with unknown variant of EDS. So I may be rarer than rare! Once you get a rare condition, others may follow. EDS is known to have comorbities – MCAS and POTS are normal. MG is rare in the general population, probably less rare with EDS patients (there’s actually a FB group for EDS & MG). As the neurologist said, EDS patients also tend to accumulate autoimmune diseases. I’ve seen reports of multiple sclerosis, rheumatoid arthritis, thyroid conditions and more. Most of us have more than one condition – often more than 3.
I’ve been fortunate not to have worse problems, to be 70 and finally reaching this stage. But finding oneself in twilight again takes a lot of adjustment.
I’ve been here before – before diagnosis.